Epilepsia la copii si adolescenti
©
Autor: Dr. Dolfi Alexandra
- Epidemiologie
- Etiologia epilepsiei la copii și adolescenți
- Factorii genetici
- Fiziopatologie
- Crizele epileptice la copii și adolescenți - Clasificare
- Diagnostic de epilepsie la copii și adolescenți
- Sindroamele epileptice la copii și adolescenți
- I. Sindromul West (spasmele epileptice infantile)
- II. Epilepsia cu absențe juvenile
- III. Sindromul Lennox- Gastaut
- IV. Absențele tipice ale copilăriei
- V: Sindromul Janz (epilepsia mioclonică juvenilă)
- VI. Epilepsia Rolandică (Parțială Benignă cu Vârfuri Centro-Temporale)
- VII. Epilepsia grand mal de trezire
- Statusul epileptic
- Principii de tratament în epilepsie la copii și adolescenți
- Tratamentul chirurgical
- Prognosticul epilepsiilor
Termenul „epilepsie” provine din grecescul „epilambaneim” care înseamnă surprindere. Astfel se încearcă explicarea aspectului imprevizibil și paroxistic pe care îl îmbracă criza epileptică.
Epilepsia este o entitate extrem de complexă care cuprinde peste 40 de sindroame, fiecare cu anumite particularități biochimice, anatomice și fiziologice care determină apariția crizelor recurente. Crizele epileptice manifeste din punct de vedere clinic se produc din cauza unor descărcări paroxistice, excesive și hipersincrone ce au loc la nivelul unui grup de neuroni cerebrali. Aceste descărcări au și un corespondent electric, putând fi detectate cu ajutorul electroencefalogramei (EEG).
Crizele pot fi:
- fie localizate la un anumit focar neuronal
- fie se pot propaga către alte arii ale creierului cuprinzând toată scoarța cerebrală și devenind generalizate.
Criza epileptică semnifică alterarea paroxistică a funcției neurologice cauzată de o descărcare electrică anormală a neuronilor cerebrali. Termenul de criză epileptică este folosit cu un sens mai larg, fiind utilizat în principal pentru diferențierea unei crize cauzate de o descărcare neuronală anormală de un fenomen paroxistic non-epileptic precum sincopa sau convulsiile febrile.
Apariția unei crize epileptice la un pacient nu pune diagnosticul de epilepsie, întrucât epilepsia este o boală cronică, caracterizată apariția mai multor crize epileptice spontane și recurente. Epilepsia nu este o boală în sine ci expresia unei disfuncții cerebrale. Crizele produse de factori reversibili precum febra, hipoglicemia, traumatismele cranio-cerebrale nu semnifică existența epilepsiei, întrucât sunt de obicei singulare și se remit spontan, intrând în categoria crizelor epileptice secundare.
Sindromul epileptic este un grup de caracteristici clinice fiind caracterizat prin: tipul crizei, vârsta de debut, aspectele electroencefalografice, factorii precipitanți ai crizelor, antecedentele heredocolaterale, evoluția în timp a bolii, prognosticul și răspunsul la medicamentele antiepileptice.
Epidemiologie
Prevalența globală a epilepsiei este între 0,5 și 0,8%, existând aproximativ 1 epileptic la 200 de locuitori. Incidența medie a epilepsiei este de 48,7 la 100.000 de locuitori, fiind diagosticat 1 caz nou pe an la 2.000 de locuitori. Cele mai multe epilepsii debutează la vârsta copilăriei, 50% fiind diagnosticate înainte de 10 ani. Epilepsia este mai frecventă la băieți, fiind mai frecvente crizele epileptice parțiale decât cele generalizate. La vârsta copilăriei, epilepsia este a doua suferință neurologică după cefalee.Etiologia epilepsiei la copii și adolescenți
Epilepsia este determinată de o interacțiune între factorii genetici și factorii dobândiți (leziuni cerebrale microscopice sau macroscopice, malformații congenitale, infecții, tumori, boli vasculare, traumatisme, intoxicații sau boli degenerative). Dacă predomină factorii genetici epilepsia este idiopatică, iar dacă predomină factorii dobândiți epilepsia este simptomatică. Este foarte important ca, în momentul diagnosticului epilepsiei să se ia în calcul cauzele posibile în raport cu vârsta de debut a crizelor, prin urmare:În perioada prenatală
Principalele cauze care ar putea duce la apariția epilepsiei sunt:- malformațiile cerebrale, date cel mai frecvent de tulburările de migrare neuronală, disgeneziile cerebrale, sindroamele neurocutanate (scleroză tuberoasă, neurofibromatoză și angiomatoza leptomeningeală caracteristică sindromului Sturge Weber) precum și malformațiile: schizencefalia, lisencefalia, pahigiria, microgiria, agenezia de corp calos și chistele arahnoidiene.
- accidentele vaculare cerebrale din viața intrauterină
- infecțiile intrauterine care afectează sistemul nervos central: toxoplasmoza, infecția cu citomegalovirus, rubeola și infecția cu herpes virus tip 2
- intoxicațiile medicamentoase sau cu toxice materno-fetale
În perioada neonatală
- encefalopatia hipoxic-ischemică determinată de nașterile distocice sau ca urmare a hipoxiei dată de tulburările cardio-respiratorii
- hemoragiile intracraniene spontane sau determinate de traumatismul obstetrical perinatal
- infecțiile cerebro-meningeene bacteriene (abces cerebral) sau virale (encefalita herpetică)
- tulburări metabolice netratate ale nou-născutului (hipoglicemie, hipocalcemie, hipoamoniemie)
În cazul în care acești factori cauzatori ai crizelor determină leziuni cerebrale severe, crizele vor apărea precoce, având caracter predominant generalizat. În cazul în care leziunile provocate de factorii cauzatori sunt discrete, crizele apar mai târziu și au în general un caracter parțial.
În perioada postnatală
- infecțiile acute precum encefalitele și meningoencefalitele cu streptococ hemolitic și streptococ pneumoniae sau cronice cu TBC sau neurosifilis. Aceste infecții determină un risc mic de epilepsie, însă acest risc crește în cazul în care crizele apar în perioada acută a infecției. Aproximativ 10% dintre copii fac epilepsie după meningită și 20% după encefalită.
- factorii imunologici sunt asociați într-o oarecare măsură cu epilepsia însă nu există studii concludente în acest sens, dar se presupune că ar putea exista un defect imunologic determinat genetic care ar putea fi subiacent epilepsiei.
- traumatismele cranio-cerebrale sunt și ele într-un număr redus de cazuri cauza epilepsiei. Crizele pot fi imediate survenind în primele secunde sau minute de la traumatism, precoce survenind în primele săptămâni post traumatism sau tardive survenind după luni, ani, chiar 10 ani de la traumatism. Această evoluție în timp depinde de perioada necesară formării leziunilor epileptogene. Crizele precoce sunt de obicei izolate, se remit spontan, însă pot fi un predictor al unei posibile viitoare epilepsii posttraumatice tardive.
Riscul dezvoltării unei epilepsii după un traumatism cranian depinde de anumiți factori precum vârsta, forța loviturii, tipul leziunii, localizarea și durata modificării conștienței. Prin urmare riscul de dezvoltare a unei epilepsii posttraumatice tardive crește în cazul fracturilor craniene înfundate cu dilacerarea substanței cenușii, în cazul hematoamelor intracraniene și a amneziei post-traumatice care depășește 24 de ore. Teoretic, lezarea oricărei zone a creierului poate determina convulsii însă potențialul epileptogen al unei leziuni este cu atât mai mare cu cât aceasta se apropie de ariile cerebrale implicate în motilitate. Anterior traumatismului poate exista și o predispoziție genetică spre convulsii astfel încât, prin adăugarea factorului de mediu constituit de leziunea cerebrală se poate dezvolta în acea zonă un focar epileptic. În practică există însă, din păcate o tendință generală de a atribui epilepsia unui traumatism cranio-cerebral din antecedente însă, trebuie ținut cont de faptul că epilepsia post traumatică apare într-un număr foarte mic de cazuri și trebuie eliminate toate celelalte cauze posibile ale convulsiilor.
- tumorile cerebrale constituie, ca și traumatismele cauze rare de epilepsie la copil, apărând în doar 1-2 % din cazuri. Mai frecvent generează epilepsie tumorile cerebrale supratentoriale mai apropiate de cortex (situate predominant în lobul frontal și temporal).
- bolile cerebrovasculare sunt foarte rare la copil, însă pot apărea crize epileptice datorate hemoragiilor spontane cauzate de ruperea malformațiilor vasculare congenitale.
- factorii toxici sunt cauze mult mai frecvente de epilepsie, întrucât aproape toate substanțele toxice produc convulsii, mai ales când este vorba de intoxicațiile acute care determină comă toxică. Pot apărea crize și în momentul în care se suprimă brusc anumite substanțe medicamentoase administrate cronic, cum sunt barbituricele și sedativele. Ingestia accidentală de alcool produce însă cel mai frecvent crizele epileptice la copii, alcoolul fiind responsabil pentru 6% dintre epilepsii.
Factorii genetici
Predispoziția către epilepsie este dată de pragul epileptogen și anume de pragul de excitație de la care neuronii încep să se descarce anormal și cu o frecvență foarte crescută. În populația sănătoasă, pragul epileptogen este suficient de crescut astfel încât stimulii obișnuiți să nu provoace descărcări neuronale haotice. La un epileptic însă, pragul epileptogen este scăzut sau foarte scăzut, astfel încât stimuli minori precum ingestia unor cantități mici de alcool sau privitul la televizor pot determina o criză epileptică. Pragul epileptogen este o caracteristică familială, cu mod variabil de transmitere. În 70- 75% din cazurile de epilepsie nu se poate găsi cauza, iar 30- 35% dintre epilepsii au un determinism genetic. Există două modalități prin care genetica intervine în epilepsie și anume:- Ereditatea mendeliană sau monogenică cu transmitere autozomal dominantă determină 1% dintre epilepsii și anume acelea din cadrul neurofibromatozelor cum ar fi scleroza tuberoasă (boala Bourneville). Expresivitatea clinică a acestora este dependentă de vârstă. Există un alt grup de epilepsii care au ereditate mendeliană monogenică cu transmitere autozomal recesivă, acesta fiind grupul epilepsiilor mioclonice progresive.
- Ereditatea multifactorială este aceea în care intervin mai multe gene diferite, care, pentru a se exprima fenotipic au nevoie de intervenția factorilor de mediu. Prin urmare, în cauzl unei leziuni cerebrale similare, numai o parte din cei afectați devin epileptici, aceștia având o predispoziție convulsivantă ereditară.
Fiziopatologie
Mecanismul de bază al epileptogenezei este existența unei populații neuronale hiperexcitabile și hipersincrone care apare constituțional sau dobândit, întrucât bazele fundamentale ale epileptogenezei sunt hiperexcitabilitatea și hipersincronia. Descărcările epileptice apar ca rezultat al unei depolarizări neuronale excesive și prelungite care generează potențiale de acțiune. Factorii epileptogeni cunoscuți până la ora actuală sunt:- Proprietățile intrinseci ale membranelor neuronale care au pe suprafața lor canale de sodiu, potasiu și calciu, a căror distribuție membranară este supusă variațiilor fiziologice și patologice ale mediului intern. Proprietățile membranare sunt responsabile pentru pragul de excitabilitate al neuronilor.
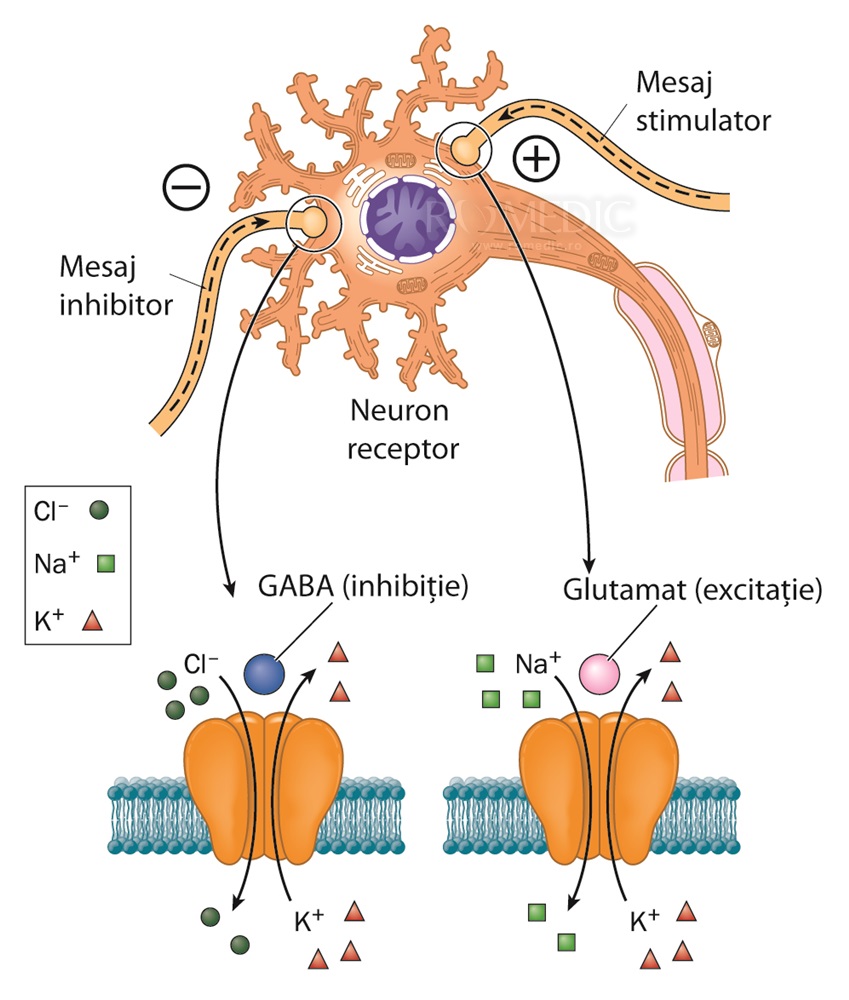
- Transmisia sinaptică este formată din sisteme excitatoare care folosesc acidul glutamic ca neurotransmițător și sisteme inhibitorii care folosesc GABA (acid gama amino butiric) ca neurotransmițător.
Procesul epileptic este datorat ruperii echilibrului între sistemele excitatorii și inhibitorii fie prin diminuarea inhibiției GABA-ergice, fie prin întărirea transmisiei excitatorii glutamat-ergice. În cazul epilepsiilor lezionale mai mulți factori intră în calcul: scăderea pragului de excitabilitate datorată remanierii histologice și funcționale creată prin redistribuția canalelor ionice, repartiția anormală a unor canale ionice și a arhitecturii lor și crearea unor noi sinapse și circuite neuronale.
La epileptogeneza primară se poate asocia epileptogeneza secundară, consecință a crizelor și leziunilor determinate de fenomene biochimice excitotoxice (moartea unor neuroni, crearea unor noi sinapse și noi circuite recurente). Se va modifica astfel relația neuron-nevroglie declanșându-se fenomenul de potențializare de lungă durată care va determina creșterea eliberării de aminoacizi excitatori.
Crizele epileptice la copii și adolescenți - Clasificare
Există două mari categorii de crize: parțiale și generalizate.În crizele generalizate descărcarea neuronală paroxistică implică simultan cortexul ambelor emisfere cerebrale. Apar tulburări de constiență și manifestări motorii bilaterale. Clinic și paraclinic nu se poate determina un semn de localizare, întrucât electroencefalograma este caracterizată prin descărcâri de vârfuri, cicluri vârf-undă sau polivârf-undă bilaterale și simetrice în toate derivațiile.
În crizele parțiale descărcarea neuronală paroxistică este limitată la un anumit sector din structurile corticale numit focar epileptic. Aspectul clinic al crizelor variază foarte mult, întrucât el depinde de localizarea focarului (temporal, parietal, frontal, occipital). Manifestările electroencefalografice sunt unilaterale și focale. Criza focală se poate însă generaliza în una sau ambele emisfere.
Din punct de vedere semiologic, crizele parțiale se împart la rândul lor în:
- crize parțiale simpe în cadrul cărora nu apare modificarea stării de conștiență
- crize parțiale complexe care apar cu modificarea conștienței fie de la început sau după un debut cu criză parțială simplă.
Aspectul simptomelor de debut ale crizelor parțiale are foarte mare importanță pentru stabilirea diagnosticului topografic. Crizele parțiale cu generalizare secundară rezultă din propagarea activității epileptice care se exprimă inițial cu simptomatologie de crize parțiale simple sau complexe și care antrenează ulterior o pierdere totală a conștienței cu simptome bilaterale, legate de difuziunea descărcării în emisferul contralateral.
Crizele epileptice generalizate
Sunt de mai multe tipuri, în funcție de modul de prezentare clinic și electroencefalografic:1. Absențele tipice sunt crize epileptice caracterizate prin pierderea bruscă a conștienței. Copilul are privirea fixă și se oprește brusc din activitate. Pot apărea și clonii papebrale sau ale capului cu durată de 5- 30 de secunde. Pe EEG apar complexe vârf-undă de 3 cicli pe secundă, bilaterale, sincrone cu debut și sfârșit brusc pe un traseu de fond normal.
2. Absențele atipice sunt caracterizate printr-un debut și sfârșit progresiv, spre deosebire de cele tipice care încep și se termină brusc, având o durată de 20- 60 de secunde și o alterare a stării de conștiență mai puțin profundă decât în cazul absențelor tipice. Apar semne asociate precum căderea capului, a trunchiului, clonii asimetrice și fenomene vegetative. Pe EEG apar complexe vârf-undă sub 3 cicli pe secundă și vârfuri care încep și se sfârșesc progresiv. Activitatea de fond este anormală, spre deosebire de absențele tipice care apar pe un fond EEG normal.
3. Crizele mioclonice sunt caracterizate prin secuse musculare masive, scurte, bilaterale și simetrice ale membrelor și trunchiului care pot determina cădere. Pierderea conștienței nu apare obligatoriu. EEG se prezintă sub forma descărcărilor polivârf-undă bilaterale, sincrone cu secusele musculare.
4. Crizele clonice sunt variante ale crizelor tonico-clonice și constau în secuse musculare clonice bilaterale, frecvent asimetrice care încetinesc progresiv și au durată variabilă. Pe EEG se observă descărcări polivârf –undă și vârf-undă bilaterale, neregulate.
5. Crizele tonice sunt caracterizate printr-o contracție musculară susținută și difuză, cu durată de secunde sau chiar minute, fiind asociată cu tulburări vegetative și alterarea stării de conștiență. Interesează de obicei musculatura extremității cefalice și mușchii axiali ai centurilor și membrelor. Pe EEG se observă polivârfuri.
6. Crizele atone au drept caracteristică diminuarea sau abolirea bruscă a tonusului muscular ceea ce determină o cădere deseori traumatizantă. Durata crizei este variabilă iar pe EEG apar vârfuri lente, neregulate.
7. Crizele tonico-clonice generalizate sau grand mal debutează brutal prin pierderea conștienței, după care urmează trei faze:
- faza tonică, cu o durată de 10- 20 de secunde constând într-o contractură susținută și generalizată, inițial în flexie apoi în extensie, însoțită de blocaj respirator și tulburări vegetative, fiind posibilă și mușcarea limbii. Pe EEG apare o activitate în ritmuri rapide de amplitudine crescândă, cu caracter bilateral, generalizat în ambele emisfere.
- faza clonică sau convulsivă este caracterizată printr-o alternanță de 30- 40 de secunde de contracții și relaxări musculare care determină secuse bilaterale bruște, intense ce devin din ce în ce mai rare apoi se opresc brusc. Sunt însoțite de apnee, cianoză, hipersecreție bronșică și respirație stertoroasă.
- faza postcritică sau stertoroasă durează de la câteva minute la câteva ore urmează imediat după faza clonică și este caracterizată printr-un somn profund sau chiar comă, hipotonie, relaxare sfincteriană, respirație amplă, zgomotoasă. Treptat se va trece la obnubilare (confuzie) care se ameliorează progresiv. Se pot asocia uneori automatisme motorii și acte agresive cu stări de agitație care pot dura câteva minute.
Crizele epileptice parțiale
Pot fi crize parțiale simple caracterizate prin păstrarea integrității stării de conștiență și crize parțiale complexe caracterizate prin alterarea conștienței. Crizele parțiale simple sunt la rândul lor de mai multe feluri:a. Motorii, având originea în cortexul motor prerolandic contralateral, corespunzând reprezentării somatotopice a homuncusului motor. Cele mai frecvente sunt crizele parțiale motorii localizate la nivelul membrului superior cu extensie progresivă și crizele versive care constau în devierea ochilor și a capului.
b. Somato-senzitive cu origine în aria postrolandică somestezică contralaterală care se manifestă prin parestezii, furnicături, senzație de descărcare electrică ce se pot propaga.
c. Senzoriale având simptomatologie diferită în funcție de focar: scotoame, hemianopsii sau halucinații (simptomatologie vizuală cu corespondență în lobul occipital), mirosuri neplăcute sau plăcute care nu există în realitate (crize olfactive cu origine în lobul temporal), senzația unor gusturi dezagreabile (crize gustative cu originea în operculul rolandic), halucinații auditive, bâzâituri, șuierat (crize auditive cu origine în lobul temporal, aria auditiva).
d. Vegetative având simptome precum paloarea, roșeața, hipertermia, hipersalivația, palpitațiile au originea în porțiunea internă a lobului temporal, în special la nivelul sistemului limbic.
e. Psihice având manifestări variate: afazie, amnezie, senzații de deja vu, sau deja vecu, sentimente stranii, stări de reverie, idei parazite, viziuni panoramice foarte rapide ale evenimentelor din trecut, crize de anxietate sau senzații plăcute, majoritatea având origine în lobul temporal.
Crizele parțiale complexe sunt caracterizate prin alterarea inițială sau secundară a conștienței, având o durată de 30 de secunde până la 2-3 minute, fiind însoțite de amnezie postcritică. Pot debuta în orice regiune corticală, dar implică cel mai frecvent lobul temporal (60% din cazuri) fiind urmat de lobul frontal. Apar cel mai frecvent ca automatisme gestuale simple sau complexe precum număratul banilor, încheiatul nasturilor, scărpinat, cătuarea unui obiect sau tendința de a pleca sau fugi. Sunt foarte frecvente și automatismele orofaringiene precum mestecat, supt și salivație.
În cazul crizelor parțiale, EEG-ul este foarte variabil. Pot să nu existe deloc anomalii sau apar descărcări de amplitudine crescută cu vârfuri și complexe vârf-undă lente, a căror topografie depinde de sediul în care are lco descărcarea epileptică.
Diagnostic de epilepsie la copii și adolescenți
Necesită parcurgerea mai multor etape. Întâi se încearcă identificarea tipului de criză, epileptică sau neepileptică. În cazul în care este vorba despre o criză epileptică, ea trebuie încadrată în una dintre tipurile de crize prezentate anterior. În funcție de vârstă, tipul de manifestare, clinică, aspectul EEG critic și intercritic, examenul obiectiv și examenul neurologic la care se adaugă stabilirea gradului de dezvoltare neuropsihică se va încerca încadrarea tabloului clinic într-un sindrom epileptic. Este foarte importantă și determinarea etiologiei crizelor, alături de stabilirea factorilor precipitanți.Pentru diagnosticul tipului de criză care este primul pas decisiv în asistența epilepsiei se va lua o anamneză detaliată centrată pe evenimentul critic. Întrebările pe care examinatorul și le pune sunt umătoarele: Episodul a fost o criză sau un alt fenomen paroxistic neepileptic? Dacă a fost o criză, a existat un eveniment acut declanșator? Criza a fost focală sau generalizată? Dacă criza a fost focală, a fost însoțită de alterarea stării de conștiență? Criza a apărut la un copil normal anterior sau la un copil cu tare neurologice? Se încearcă și stabilirea duratei, frecvenței, ritmului circadian, aspectului stereotip monomorf sau polimorf și contextul în care au apărut crizele.
De o foarte mare importanță este vizionarea directă de către examinator a crizei sau înregistrarea video la domiciliu. De aceea, în cazul unei anamneze și a unui examen clinic neconcludent, când nu s-a putut stabili natura sau tipul crizei, părinții sunt sfătuiți să realizeze o înregistrare video la domiciliu a fenomenelor convulsive ulterioare. Foarte utilă este și înregistrarea video- EEG întrucât permite stabilirea diagnosticului corect clinico-EEG de criză epileptică.
Examinarea neurologică în timpul crizei sau imediat după criză va permite evidențierea anumitor simptome neurologice de focar, lateralizate sau localizate care pot da indicii prețioase cu privire la sursa focarului epileptic. Examenul neurologic va căuta de asemenea elemente de suferință ale sistemului nervos cu caracter acut sau cronic, simptome ce pot fi progresive, stabilizate sau sechelare. Foarte important este faptul că o criză epileptică poate determina simptome neurologice tranzitorii și chiar tratamentul antiepileptic poate induce simptome și semne neurologice.
Examenul obiectiv va fi atent și riguros, întrucât poate evidenția anumite simptome fizice în relație cu suferința sistemului nervos central.
La sfârșitul examinării se va face o sesiune de hiperventilație timp de 3-4 minute. Hiperventilația declanșează de obicei crizele de absență de aceea este un element prețios de diagnostic al crizelor de absență la un copil netratat.
Evaluarea psihologică este indispensabilă întrucât statusul psihic al copilului este o cerință a protocoalelor internaționale de stabilire a diagnosticului de sindrom epileptic. Vor fi evaluate funcțiile cognitive, afectiv-emoționale și comportamentale ale copilului. Evaluarea se va face la debutul bolii epileptice și pe parcursul evoluției bolii întrucât tratamentul antiepileptic este potențial cauzatoare a unor disfuncții psihice în anumite cazuri, determinând cel mai frecvent tulburări de adaptare care sunt tratabile.
Trebuie reținut faptul că EEG are un rol esențial în diagnosticul epilepsiei atunci când înregistrează anomalii specifice diferitelor forme clinice, însă numai modificările EEG singure, în absența corespondentului clinic nu pot stabili singure diagnosticul.
Diagnostic diferențial
Se face în raport cu tipul crizei. Astfel, în cazul crizelor tonico-clonice generalizate se va face cu sincopa, spasmul hohotului de plâns, crizele psihogene, cataplexia, hipoglicemia și bolile cardiovasculare.În cazul crizelor parțiale simple se va realiza cu accidentul ischemic tranzitor, mișcările anormale (ticuri, distonii), mișcările ritual ede balansare a capului și trunchiului (întrerupte când copilul este strigat pe nume), migrena, tulburări motorii de origine toxică produse de unele medicamente și tetania.
Crizele parțiale complexe trebuie deosebite de parasomnii (somnambulism), coșmar, automatisme masticatorii (bruxism = scrâșnit din dinți), tulburările paroxistice de comportament (atacuri de panică, stări crepusculare) și enurezis.
Teste paraclinice
Electroencefalograma este cea mai importantă investigație paraclinică în cazul epilepsiei. Oferă informații pentru susținerea diagnosticului clinic și ajută la clasificarea epilepsiei în generalizată sau focală, uneori chiar la stabilirea unui tip aparte de sindrom epileptic. Ajută și la identificarea structurilor subiacente focarului epileptic, la supravegherea pacientului în evoluție, la supravegherea tratamentului sau la deciderea opririi tratamentului. Însă deși unii pacienți sunt cert epileptici (fapt stabilit clinic) EEG-ul lor nu surprinde activitarea neuronală paroxistică, acest lucru întâmplându-se în 50% din cazuri. Prin urmare, EEG singur nu stabilește niciodată diatnosticul în lipsa certificării clinice a crizei epileptice (fie prin vizualizarea acesteia direct de către examinator fie prin vizionarea înregistrărilor video realizate de către părinți acasă). EEG poate arăta și trasee caracteristice ale unor boli neurodegenerative progresive care pot fi cauze ale epilepsiei. Se folosește și pentru monitorizarea răspunsului la tratament, mai ales în cazul absențelor tipice și în sindromul West.Probele de sensibilizare sunt obligatorii pentru orice înregistrare EEG, acestea constând în:
- hiperventilație care produce hipocapnie (scăderea concentrației de dioxid de carbon din sânge) ceea ce determină scăderea debitului sanguin cerebral cu declanșarea crizei epileptice, în principal în cazul crizelor de absență.
- stimularea luminoasă intermitentă declanșează convulsiile mai ales la acienții care suferă de epilepsie generalizată idiopatică
- EEG de somn, de preferat pe parcursul nopții. Este foarte important întrucât somnul este un activator eficace al anomaliilor intercritice (care apar în perioada dintre crizele epileptice), în special în epilepsiile idiopatice. Uneori stabilirea diagnosticului se poate face numai pe baza EEG de somn, cum este în cazul epilepsiilor parțiale idiopatice cu paroxisme rolandice. Activarea prin somn este utilă și când se suspicionează epilepsia benignă cu vârfuri centro-temporale.
- Identificarea modificărilor intercritice trebuie făcută foarte atent întrucât 10-15% din populația generală poate avea anomalii nespecifice minore, iar 1% au anomaliis pecifice de vârfuri sau polivârfuri generalizate sau focale, în lipsa simptomatologiei clinice. Între 5 și 45% dintre rudele de gradul I ale unui pacient cu crize primar generalizate vor avea descărcări de complexe vârf-undă de 3 cicli pe secundă deși nu au avut niciodată crize epileptice. Pot fi întâlnite anomalii epileptiforme la 30% dintre persoanele cu tulburări de învățare.
Computer-Tomografia cerebrală
Are ca indicații identificarea cauzei crizelor (identificarea leziunii), evidențierea calcificărilor cerebrale și excluderea tumorilor cerebrale. Dacă CT-ul inițial este normal, pentru a se identifica eventuale anomalii evolutive (de exemplu o tumoră cerebrală cu creștere lentă) CT se poate repeta după luni sau ani. Orice criză inaugurală fără etiologie clară impune realizarea unui CT cerebral.RMN cerebral
Este superior CT-ului întrucât poate da informații suplimentare când CT este normal evidențiind mult mai bine displaziile difuze, leziunile vasculare, demielinizările și leziunile specifice precum hamartoamele. Este examenul de primă intenție în epileptologie, mai ales în epilepsiile refractare. Este total inofensiv (nu presupune iradiere) și este mult mai sensibil pentru detectarea leziunilor focale și a micilor malformații. Este de asemenea și mult mai scump.Alte explorări
Radiografia simplă de craniu poate identifica calcificări patologice intracraniene (tumori, facomatoze, parazitoze) și sechelele osoase ale traumatismelor cranio-cerebrale.Angiografia se justifică numai în explorarea malformațiilor arterio-venoase cerebrale.
Explorările izotopice precum PET (Tomografia cu emisie de pozitroni) și SPECT (tomografia cu emisie monofotonică) sunt realizate în chirurgia epilepsie pentru realizarea bilanțului prechirurgical al epilepsiilor parțiale cu delimitarea precisă a focarului care va fi inactivat chirurgical. Ele măsoară activitatea metabolică cerebrală locală în criză care se manifestă prin creșterea consumului local de glucoză și oxigen și creșterea debitului sanguin cerebral regional și intercritic (hipometabolism cu hipodebit cerebral).
Ecografia trans-fontanelară are limită strict în perioada neonatală pentru identificarea hemoragiilor cerebrale la nou-născut, a malformațiilor și a eventualelor neoformații și calcificări. Se realizează la sugar și copilul mic atât timp cât este deschisă fontanela anterioară.
Paraclinic, în cazul suspicionării unor etiologii specifice mai sunt necesare și: bilanțul inflamator, bilanțul imunologic, dozarea acizilor organici, fundul de ochi și prelevările bioptice (piele, mușchi).
Monitorizarea video-EEG înregistrează continuu comportamentul și EEG-ul unor copii epileptici surprinzând criza clinic corelat cu manifestarea electrică a descărcărilor neuronale.
Sindroamele epileptice la copii și adolescenți
Din punct de vedere etiopatogenic există trei mari clase de epilepsii:- Idiopatice care survin independent de orice leziune cerebrală, investigațiile neuroimagistice de structură fiind negative. Factorul principal este predispoziția ereditară reală sau presupusă (care nu se poate demonstra).
- Simptomatice care sunt rezultatul unei leziuni difuze sau locale, evolutive sau fixe a sistemului nervos central. Această leziune poate fi obiectivată printr-un deficit neurologic și clinic și o anomalie biologică evidențiabilă cu ajutorul explorărilor paraclinice.
- Criptogenice (au cauză ascunsă). Ele sunt presupuse a fi simptomatice, însă leziunea de bază scapă mijloacelor de investigație în momentul dat. Investigațiile se vor repeta astfel după o perioadă mai lungă de timp. O epilepsie criptogenică cu CT normal poate apărea ca simptomatică în urma unui RMN.
Principalele sindroame epileptice la copii și adolescenți sunt clasificate în funcție de criteriul vârstei de apariție (în funcție de maturarea cerebrală). Ele sunt următoarele:
I. Sindromul West (spasmele epileptice infantile)
Acest sindrom este caracterizat prin existența unei triade simptomatologice: spasme infantile (o formă particulară de crize epileptice), oprirea sau regresia dezvoltării psihomotorii din momentul apariției spasmelor, hipsaritmie pe EEG (traseu EEG specific- descărcări de unde rapide și ample). Sindromul West survine aproape întotdeauna în primul an deviață, cu o incidență de 24- 42 la 100.000 de nașteri, băieții fiind mai frecvent afectați decât fetele.Clinic
Spasmele infantile sunt contracții musculare bruște, bilaterale și simetrice, tonice sau clonice. Cel mai frecvent apare flexia bruscă a capului, trunchiului și membrelor asemănător cu un briceag care se închide.Membrele sunt flectate în abducție sau adducție, spasmele producându-se rar în extensie. Cele două tipuri de spasme infantile pot coexista sau se pot succeda la același sugar. Intensitatea spasmelor variază în timp și de la un spasm la altul, uneori limitându-se la o simplă cădere a capului, o fixare a privirii sau doar la un plâns inexplicabil. Apar foarte frecvent în salve de număr variabil (în cazurile grave chiar peste 200) separate între ele de un interval de 5- 30 de secunde. Intensitatea spasmelor poate crește sau diminua în cursul aceleiași salve. Se poate asocia uneori și o pierdere de conștiență greu perceptibilă. La sfârșitul salvei apar mișcări oculare anormale, modificări vasomotorii (paloare, roșeață), grimase sau surâs. Pe durata salvei sugarul poate fi aton sau din contră hiperton. Salva de spasme apare de obicei după somn, la trezire sau la adormire, foarte rar în timpul somnului lent. De obicei după vârsta de un an o dată cu maturizarea cerebrală spasmele dispar, rareori pot persista încă câțiva ani. Cu timpul însă se pot adăuga alte tipuri de crize, parțiale, atone, tonice dar acestea apar în cazul în care spasmele infantile au avut o etiologie simptomatică.
Într-un procent de 25% spasmele infantile determină încetinirea dezvoltării psihomotorii și/sau regresul achizițiilor. Sugarul își pierde treptat interesul pentru obiecte, pierde prehensiunea voluntară, surâsul și urmărirea oculară iar controlul postural se deteriorează. Într-un procent de 5% dezvoltarea psihomotorie este absolut normală, prognosticul fiind foarte bun în acest număr redus de cazuri.
Aspect electroencefalografic
Este foarte heterogen, cel mai des fiind caracterizat prin hipsaritmie, care este un amestec haotic de unde lente foarte ample cu unde ascuțite și vârfuri de amplitudine și topografie variabilă, cu activitate asincronă între cele două emisfere cerebrale, în absența ritmului normal de fond. În somnul lent anomaliile se fragmentează în vârfuri și complexe vârf-undă lente difuze și sincrone separate de trasee cu amplitudine scăzută.Etiologie
70-80% dintre spasmele infantile sunt simptomatice, fiind consecințe ale unor encefalopatii hipoxic-ischemice perinatale, malformații cerebrale, scleroză tuberoasă, infecții postnatale, tulburări metabolice sau factorilor preexistenți debutului spasmelor. 50% din cazuri sunt datorate unui fenomen pur funcțional fiind numite spasme infantile benigne în timp ce 15% din spasmele infantile au o etiologie criptogenică.Tratament
Spasmele infantile sunt în general rezistente la toate antiepilepticele. Cel mai aplicat este tratamentul cu ACTH (hormon adenocorticotrop hipofizar) în doze de 20 unități pe zi care se vor crește până la 40 unități pe zi, uneori chiar până la 160 unități pe zi. Este menținută aceeași doză până când dispar spasmele și hipsaritmia după care cantitatea administrată se reduce treptat până la scoaterea completă a medicamentului. Dozele mari nu au efect superior față de cele moderate sau mici, însă efectele secundare sunt mai importante la dozele mari de aceea se folosește ACTH sintetic sub denumirile Synacten sau Cortrosyn în doză de 0,1 mg/kg corp pe zi la care se adaugă prednison 2-3 mg/kgcorp/zi sau hidrocortizon hemisuccinat 5-20 mg/kgcorp/zi. Combinația dintre ACTH și corticosteroizi a arătat efecte superioare ACTH administrat singur în studiile clinice. Tratamentul se începe cât mai precoce în vederea ameliorării prognosticului.Corticoterapia are însă numeroase efecte secundare precum obezitatea cushingoidă, încetinirea creșterii, hipertensiune arterială, infecții, osteoporoză, alcaloză hipokaliemică și alte tulburări electrolitice, miocardiopatie hipertrofică. Și ACTH are efecte secundare prin urmare este necesară o monitorizare permanentă a ionogramei și a tensiunii arteriale.
În afară de corticoterapie și ACT se mai pot adăuga:
- Benzodiazepine- clonazepam maximum 6 mg pe zi, sau nitrazepamul (cea mai folosită benzodiazepină în sindromul WEST) care poate determina dispariția spasmelor în aproximativ 2 săptămâni. Nitrazepamul este considerat alterantiva la tratamentul cu ACTH la copiii la care ACTH nu poate fi utilizat din cauza efectelor secundare.
- Acidul valproic este eficient în doze mari în aproximativ 45% dintre spasmele epileptice infantile însă trebuie ținut cont de potențialul său toxic. Trebuie folosite întâi benzodiazepinele și corticoterapia și dacă acestea nu au efect trebuie adăugat acidul valproic la schema de tratament.
- Vigabatrinul este foarte eficient în spasmele epileptice simptomatice, mai ales în cele asociate cu scleroza tuberoasă. Doza este de 100-150mg/kg/zi administrat singur sau asociat cu alte antiepileptice.
- Piridoxina este asociată cu ACTH sau valproat în doze de 40-50mg/zi având efect neuroprotector.
- Imunoglobulinele i.v. în doze mari repetate la 2-3 săptămâni se pot adăuga la medicamentele antiepileptice.
- Zonisamidul este un nou antiepileptic folosit cu succes în sindromul West nou diagnosticat.
Prognosticul spasmelor infantile este în general sever mai ales în formele simptomatice, întrucât 70% dintre sugari rămân cu retard psihic iar 50- 60% fac alte tipuri de epilepsie în special sindrom Lennox- Gastaut. Spasmele infantile pot dispărea și spontan iar spasmele epileptice infantile benigne au un prognostic foarte bun putându-se vindeca total și permițând o dezvoltare psihică normală.
II. Epilepsia cu absențe juvenile
Este o formă de epilepsie similară cu epilepsia cu absențe tipice a copilului dar are un debut mai tardiv, între 10-17 ani cu frecvență maximă între 12 și 14 ani. Absențele apar mai puțin frecvent pe descărcările EEG, sunt mai rapide 4 cicli/secundă și se asociază frecvent cu crize tonico-clonice generalizate de trezire și cu crize mioclonice. Prognosticul nu este favorabil, aceste tipuri de crize nerăspunzând foarte bine la medicația antiepileptică.III. Sindromul Lennox- Gastaut
Constituie 1-5% din toate formele de epilepsie ale copilăriei fiind cea mai comună formă de epilepsie refractară la tratamentul antiepileptic. Debutează între 1 și 8 ani, cu maxim la 3-5 ani fiind caracterizat prin crize polimorfe, tonice, atone, mioclonice sau absențe atipice cu început și sfârșit gradat. Tipurile de crize enumerate mai sus pot fi combinate, de exemplu o criză atonă cu cădere bruscă poate fi urmată de o criză tonico-clonică.Pe EEG sunt vizualizate complexe vârf-undă lentă de 1-1,3 cicli/secundă de lungă durată.
Etiologia este de tip criptogenic sau simptomatic
Ca tratament se încearcă combinații de antiepileptice, nu întotdeauna cu rezultate bune: Valproat, Lamotrigine, Vigabatrin, Carbamazepin, Benzodiazepine, Corticoterapie, Imunoglobuline și tratament chirurgical (calosotomie- secționarea corpului calos cu disconexie interemisferică).
Prognosticul este prost, crizele se reduc treptat sub tratament însă retardul psihomotor persistă.
20% din cazurile de sindrom Lennox-Gastaut sunt precedate de sindromul West.
IV. Absențele tipice ale copilăriei
Sunt denumite în literatura veche petit mal. Sunt destul de comune, constituind peste 5% dintre epilepsiile copilăriei. Debutul are loc între 3-12 ani cu maximum la 6-7 ani. Sunt mai frecvente la fete.Crizele de absență au un început și sfârșit brusc, se pierde total contactul cu mediu pe o durată de 5-15 secunde, frecvența crizelor fiind foarte mare, chiar de 100-200 pe zi. Absențele se asociază frecvent cu clonii ale pleoapelor, automatisme gestuale simple, scurtă emisie urinară, scurtă și ușoară diminuare a tonusului postural, modificarea respirației și midriază.
Pe EEG se vizualizează complexe vârf-undă de 3 cicli/secundă cu debut și sfârșit brusc pe un traseu de fond normal.
Etiologia este idiopatică iar tratamentul constă în administrarea în monoterapie sau pluriterapia cu Valproat, Etosuccinimid și Lamotrigină (nu se administrează niciodată Carbamazepină în absențe).
Prognosticul este variabil, în 75% din cazuri crizele se remit însă în 40% din cazuri apar crizele tonico-clonice generalizate în adolescență, iar 30% dintre copii rămân cu tulburări cognitive ușoare.
O caracteristică foarte importantă este aceea că hiperventilația induce criza de absență, aceasta constituind un test diagnostic.
Epilepsia cu absențe mioclonice apare în general la băieții între 1- 12 ani având un aspect de absențe asociate cu crize generalizate mioclonice. EEG în acest caz este similar cu cel din epilepsia absență tipică dar tratamentul combinat cu Valproat și Etosuccinimidă are un efect slab, dar Lamotrigina oprește crizele. În general în acest tip de absență mioclonică dezvoltarea psihomotorie are de suferit, prognosticul fiind mai puțin bun.
V: Sindromul Janz (epilepsia mioclonică juvenilă)
Este cel mai comun sindrom epileptic al adolescenței, cu debut la 10-18 ani sau chiar mai târziu.Crizele sunt sub formă de mioclonii bilaterale singulare sau multiple cu predominență la membrele superioare. Pot antrena căderea. Frecvent sunt legate de trezire, oboseală, deprivare de somn sau vizionare TV. Se asociază în 90% din cazuri cu crize generalizate și în 10-30% din cazuri cu absențe.
EEG este normal sau apar scurte descărcări generalizate de complexe vârf-undă sau polivârf-undă neregulate de peste 3 cicli/secundă. 70-80% din cazuri sunt fotosensibile, criza fiind indusă de stimularea luminoasă intermitentă.
Etiologia este idiopatică, având un substrat genetic, antecedentele heredocolaterale de epilepsie fiind foarte frecvente, gea asociată defectului este considerată a fi situată pe cromozomul 6.
Prognosticul este bun, deoarece răspunde bine la medicamentele antiepileptice cu răspuns bun la Valproat și Lamotrigină însă epilepsia este farmaco dependentă, crizele revenind după oprirea tratamentului, de aceea este necesar tratament permanent pe toată durata vieții.
VI. Epilepsia Rolandică (Parțială Benignă cu Vârfuri Centro-Temporale)
Vârsta de debut este între 3 și 13 ani cu frecvență maximă între 7 și 9 ani fiind cea mai frecventă epilepsie parțială a copilăriei (10-15% dintre toate epilepsiile copilului).Crizele sunt parțiale senzitivo-motorii și debutează cu aură (parestezii unilaterale) după care urmează convulsii tonice și/sau clonice care cuprind treptat limba, buzele, obrazul, faringele, laringele, uneori brațul și mai rar tot corpul. Se produce hipersalivație marcată, conștiența poate fi păstrată.
EEG este caracteristic evidențiind vârfuri centro-temporale uni sau bilaterale activate de somn.
Este o epilepsie idiopatică, suspicionându-se un substrat genetic.
Crizele se remit de obicei spontan, cedând în general spre pubertate, deși inițial sunt foarte frecvente. Nu este necesar tratament obligatoriu, se poate încerca cu Carbamazepină sau Valproat dacă crizele sunt foarte frecvente, răspunsul la tratament fiind foarte bun. Prognosticul este favorabil, nu au loc întârzieri ale dezvoltării psiho-motorii.
VII. Epilepsia grand mal de trezire
Este rară, 1-5% dintre toți copiii cu epilepsie au această formă. Debutul este între 6-20 de ani cu frecvență maximă între 11-15 ani.Crizele sunt tonico-clonice generalizate după trezire sau în perioadele de relaxare. Se pot asocia cu absențe și crize mioclonice.
Pe EEG se evidențiază descărcări de complexe vârf-undă generalizate.
Etiologia este idiopatică.
Tratamentul se poate realiza cu Valproat, Carbamazepină, Lamotrigină sau fenobarbital.
Prognosticul este în general favorabil întrucât crizele răspund foarte bine la tratament dar sunt farmacodependente, reapar la întreruperea tratamentului fiind necesară medicație permanentă.
Statusul epileptic
Status epilepticus sau starea de rău epileptic este o stare de urgență care apare după o serie de crize epileptice succesive fără reluarea conștienței între crize sau ca urmare a unei activități epileptice continue, cu durata de peste 30 de minute.Statusul epileptic se poate produce la un epileptic cunoscut, poate inaugura o epilepsie sau poate fi simtomatic unei afecțiuni cerebrale acute. Poate surveni la orice vârstă, mai frecvent la copii și bătrâni.
Din punct de vedere clinic este foarte heterogen, există atâtea variante clinice de status câte crize epileptice sunt. Poate apărea în orice tip de criză, dar cel mai frecvent în crizele tonico-clonice generalizate. Există și status-absență manifestat sub forma unei stări confuzionale prelungite.
Diagnosticul de certitudine este dat de revenirea conștienței concomitent cu dispariția anomaliilor EEG după benzodiazepine intravenos. La un epileptic cunoscut, un status epilepticus iminent poate fi anunțat prin creșterea frecvenței și duratei crizelor, moment în care, dacă se intervine medicamentos se poate opri intrarea copilului în status.
Cauze de apariție a status epilepticus:
- întreruperea bruscă a tratamentului antiepileptic
- consumul de alcool
- afecțiuni intercurente
- privarea de somn
- toxice exogene și medicamente (antidepresive triciclice, neuroleptice, intoxicații cu monoxid de carbon și organofosforice)
- anoxie cerebrală acută
- afecțiuni metabolice ale sistemului nervos central
- dezechilibrul hidro-electrolitic (hiponatremie, hipocalcemie, hipopotasemie, hipoglicemie, hiperglicemie, hipernatremie)
- leziuni cerebrale acute (traumatism cranio-cerebral, accident vascular cerebral ischemic sau hemoragic)
- infecții cerebrale (meningo-encefalită sau abces cerebral)
- tumori cerebrale
Statusul epilepticus prelungit poate determina sechele foarte grave neurologice datorate leziunilor anoxo-ischemice. Leziunile sunt ireversibile când crizele durează mai mult de 90 de minute. Sechelele pot fi și somatice: renale, cardiace, hepatice, edem cerebral sau chiar moarte (4% din cazuri) prin stop cardio-respirator.
Tratamentul statusului epileptic
Statusul epileptic este o urgență medicală și neurologică iar tratamentul trebuie început cât mai devreme posibil pentru prevenirea sechelelor și a mortalității. Medicamentele se administrează în doze suficiente pe cale intravenoasă. Se va începe cu benzodiazepine i.v. sau intrarectal (Diazepam 0,4 mg/kg sau Lorazepam 0,05-0,1 mg/kg). Dacă crizele continuă doza se va repeta, însă dacă crizele nu se opresc se asociază fenitoin i.v. 20 mg/kg. Dacă crizele durează de peste 40 de minute copilul va fi transferat la serviciul de terapie intensivă pentru intubație traheală și ventilație asistată pentru a fi evitate sechelele hipoxiei. Pentru crizele care persistă peste 100 de minute se va face anestezie generală cu curarizare (Thiopental). După ce copilul a ieșit din status epilepticus se va institui urgent tratamentul de întreținere pe cale orală.Principii de tratament în epilepsie la copii și adolescenți
Tratamentul etiologic este responsabil de înlăturarea cauzelor epilepsiei: poate fi medical (corectarea unor tulburări metabolice) sau chirurgical (extirparea unor tumori, abcese, chisturi sau hematoame)Tratamentul medicamentos este instituit pe baza anumitor reguli.
Se va institui numai în cazul în care diagnosticul de epilepsie este cert, prin urmare se amână introducerea tratamentului până la certitudinea diagnosticului. Tratamentul medicamentos trebuie aplicat cât mai precoce de la debutul epilepsiei deoarece repetarea crizelor contribuie la întărirea mecanismelor patologice declanșatoare și la scăderea pragului epileptogen precum și la apariția mai precoce a sechelelor și a retardului psihomotor.
Monoterapia (administrarea unui singur antiepileptic) este modalitatea prin care debutează tratamentul atunci când epilepsia se manifestă printr-un singur tip de crize sau prin mai multe tipuri de crize toate controlabile prin același medicament. Se va trece la biterapie sau politerapie numai în momentul în care are loc eșecul monoterapiei corect administrate.
Alegerea medicamentului va fi făcută în favoarea preparatului mai puțin toxic și cu cele mai puține efecte adverse și secundare asupra funcțiilor congintive și comportamentale fiind ales și în funcție de tipul crizei. Doza de medicament variază în funcție de vârsta pacientului, de gravitatea procesului epileptic și de greutatea copilului.
Tratamentul se va introduce lent, progresiv, în 2-3 paliere de câte 2-7 zile pentru a evita efectele secundare. Dacă după perioada de acumulare a medicamentului crizele continuă și nu apar efecte adverse, se va crește doza zilnică treptat, până la dozele maxime, cu condiția absenței efectelor adverse. Administrarea rapidă a medicamentului se folosește numai în cazurile în care frecvența crizelor este mai mare decât intervalul de timp suficient pentru realizarea concentrațiilor serice stabile.
Ulterior doza se poate reduce în funcție de controlul crizelor până la limita inferioară care asigură controlul complet al crizelor. Numărul de prize zilnice depinde de farmacocinetica fiecărui antiepileptic și anume cele care se acumulează rapid pot fi administrate într-o singură priză (fenobarbital, etosuccinimidă, fenitoină), în timp ce medicamentele cu eliminare rapidă se vor administra în 3-4 prize zilnice la interval de 6-8 ore (carbamazepină, acid valproic, diazepam).
Supravegherea eficienței tratamentului medicamentos este în primul rând clinică, criteriul de eficacitate fiind absența crizelor epileptice.
Se va face monitorizarea serică a drogului pentru verificarea complianței la tratament și a interacțiunilor medicamentoase și pentru corecția tratamentului (mai ales la fenitoină, carbamazepină și fenobarbital).
Examinările clinice periodice EEG și de laborator sunt obligatorii fie pentru depistarea precoce și corecția efectelor secundare, fie pentru corectarea dozei. Unele efecte secundare pot fi ireversibile (de exemplu fenitoina produce atrofie cerebeloasă și hipertrofie gingivală).
Tratamentul trebuie menținut obligatoriu zilnic o perioadă de 2 până la 10 ani sau chiar toată viața, acest lucru depinzând de forma clinică de epilepsie. De exemplu, în epilepsia mioclonică juvenilă care este cea mai farmacodependentă formă de epilepsie tratamentul va fi menținut toată viața, altfel crizele revin.
Suprimarea bruscă, de către părinte, fără avizul medicului curant a tratamentului antiepileptic implică un risc major de reapariție a crizelor și creșterea frecvenței crizelor, copilul putând intra în status epilepticus.
Suprimarea completă a tratamentlui sub aviz medical se va face la pacienții stabilizați după 2-5 ani de control al crizelor. Descreșterea dozei în vederea scoaterii medicației se va face progresiv, pe durata a 6-18 luni prin scăderea cu 25% din doză la fiecare 6 luni. Pe perioada reducerii dozelor pacientul este sfătuit să evite factorii declanșatori ai crizelor (stimularea luminoasă intermitentă = discotecile, privitul la TV și calculator, hiperventilația, efortul prelungit, alcoolul). Reapariția modificărilor EEG paroxistice subclinice în cursul reducerii dozelor impun amânarea etapei următoare de reducere și reevaluarea copilului/adolescentului. Dacă reapare o criză se poate recrește tratamentul la o doză anterioară.
Tratamentul chirurgical
Se ia în discuție în caz de rezistență la toate medicamentele antiepileptice. Este încă în plină dezvoltare. Constă în:- Intervenții paleative care încearcă să amelioreze frecvența crizelor fără să vindece epilepsia dar împiedicând propagarea crizei. Se realizează calosotomii (în epilepsii generalizate simptomatice grave cu căderi frecvente) și hemisferectomii (în epilepsii grave cu hemiplegie ceregrală infantilă).
- Intervenții curative care încearcă să extirpe țesutul cerebral la nivelul căruia se află focarul epileptic. Acest tratament se limitează la epilepsiile parțiale rebele la tratamentul medical și cu o evoluție de cel puțin 2 ani care au un focar epileptogen situat într-o zonă cerebrală a cărei excizie nu provoacă deficit neurologic sau neuropsihic semnificativ. Se încearcă identificarea focarului epileptogen prin înregistrări epileptice cu electrozi de profunzime și prin imagerie funcțională (PET, SPECT).
Prognosticul epilepsiilor
Este privit din punctul de vedere al persistenței crizelor, al implicațiilor cognitiv-comportamentale și al mortalității. Într-un anumit număr de cazuri epilepsia dispareDin Biblioteca medicală vă mai recomandăm:
Din Ghidul de sănătate v-ar putea interesa și:
Forumul ROmedic - întrebări și răspunsuri medicale:
Pe forum găsiți peste 500.000 de întrebări și răspunsuri despre boli sau alte subiecte medicale. Aveți o întrebare? Primiți răspunsuri gratuite de la medici.- Epilepsie (sindrom West) copil 12 luni
- Epilepsia si contraceptia-doresc un raspuns de la un medic
- Lamictalul si orfirilul in timpul sarcinii?...+ operatie pe creier...
- Matusa mea are epilepsie si ia pastile, dar fara rezultat...
- Epilepsie
- Tratament adjuvant cu gemoterapice în epilepsie
- Suspect de epilepsie
- Probleme de picioare
- Alimente pentru stimularea activitatii cerebrale
- Pierderi de memorie