Sindromul Kallmann
Autor: Bezim Claudia
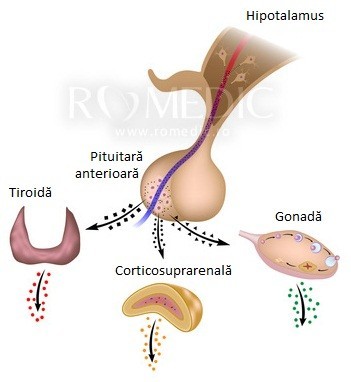
Sindromul Kallmann sau deficitul izolat de GnRH (gonadoliberina, hormonul eliberator de gonadotropină) reprezintă o afecțiune caracterizată prin pubertate întârziată sau absentă și prin lipsa simțului olfactiv (simțul mirosului). [1]
Sindromul Kallmann este o formă de hipogonadism hipogonadotrop, ceea ce înseamnă că producția de hormoni care influențează dezvoltarea sexuală este afectată. [1]
Există patru forme de sindrom Kallmann, diferențiate în funcție de cauza genetică. Toate cele patru forme sunt caracterizate de hipogonadism hipogonadotrop și de modificări ale simțului olfactiv. Alte modificări, cum ar fi despicătura palatină, apar doar în tipul 1 și 2. [1]
Cum apare sindromul Kallmann?
Sindromul Kallmann apare la 1 din 10.000 de bărbați, respectiv la 1 din 50.000 de femei. Este de cinci ori mai frecvent la sexul masculin decât la sexul feminin. Cea mai frecventă formă întâlnită este tipul 1. [3]
Sindromul Kallmann este cauzat de mutațiile anumitor gene (KAL1, FGFR1, PROKR2 și PROK2). Mutațiile genelor duc la întreruperea migrării neuronilor olfactivi și a neuronilor implicați în producerea de GnRH în timpul dezvoltării creierului. [1]
Lipsa migrării neuronilor olfactivi duce la scăderea sau chiar lipsa simțului mirosului. Lipsa migrării neuronilor implicați în producerea de GnRH duce la lipsa producerii hormonilor sexuali. Acest lucru duce la hipogonadism hipogonadotrop și la tulburări în dezvoltarea normală sexuală. [1]
Numai în 25-30% dintre cazurile de sindrom Kallmann este identificată mutația genelor respective. În anumite cazuri, mutația nu poate fi identificată, cauza sindromului nefiind necunoscută. Există ipoteza implicației altor gene în determinarea sindromului Kallmann. [1]
Genele asociate sindromului Kallmann sunt implicate în dezvoltarea anumitor zone din creier înainte de naștere. Genele respective determină formarea și migrarea unui grup de celule nervoase specializate în simțul olfactiv, adică neuronii olfactivi. Un alt rol al genelor este migrarea neuronilor care produc hormonul GnRH. GnRH controlează producția unor hormoni implicați în dezvoltarea sexuală înainte de naștere și în perioada pubertară. Acești hormoni sunt importanți pentru funcționarea normală a gonadelor (ovarele la femeie și testiculele la bărbat). [1]
Semne si simptome
Bărbații care suferă de hipogonadism hipogonadotrop sunt, de obicei, născuți cu micropenis (penisul are dimensiuni foarte mici) și criptorhidie (testiculele nu au coborât în scrot). La puberate, bărbații nu dezvoltă caractere sexuale secundare, cum ar fi creșterea părului facial și axilar sau îngroșarea vocii. [1]
Bărbații cu sindrom Kallmann prezintă:
- Libido scăzut;
- Masă musculară scăzută;
- Disfuncție erectilă;
- Infertilitate. [2]
Femeile cu sindrom Kallmann nu prezintă menstruații și nici dezvoltarea sânilor. Uneori, pubertatea poate să apară, însă este incompletă sau întârziată. [1]
Persoanele diagnosticate cu sindrom Kallmann prezintă și hiposmie (scăderea simțului olfactiv) sau anosmie (absența simțului olfactiv). Această caracteristică (modificări ale simțului olfactiv) diferențiază sindromul Kallmann de alte tipuri de hipogonadism hipogonadotrop, care nu prezintă modificări ale simțului mirosului. [1]
Frecvent, persoanele care au sindrom Kallmann nu sunt conștiente de faptul că simțul olfactiv este diminuat sau absent. Acest lucru este pus în evidență prin anumite teste. [1]
Simptomatologia sindromului Kallmann variază, chiar între persoanele din aceeași familie. [1]
Alte simptome ale sindromului Kallmann sunt:
- Agenezie renală unilaterală (lipsa dezvoltării unui rinichi);
- Despicătură labială;
- Despicătură palatină;
- Anomalii de dezvoltare ale dinților;
- Tulburări auditive, surditate;
- Degete scurte;
- Mișcări anormale ale globilor oculari;
- Obezitate;
- „Miscarea mâinii în oglinda” (mișcare involuntară a unei mâini, care imită mișcarea celeilalte mâini). Această simptomatologie poate împiedica realizarea anumitor acțiuni, cum ar fi utilizarea unui instrument muzical. [1] [3]
Deficitul simțului olfactiv și tulburările în dezvoltarea sexuală sunt explicate prin lipsa migrării neuronilor olfactivi și a celor responsabili de producerea GnRH. Având în vedere faptul că simptomatologia variază de la om la om, există ipoteza altor anomalii genetice și a implicării unor factori de mediu. [1]
Modalități de transmitere
Sindromul Kallmann 1 are transmitere legată de cromozomul X. Gena KAL1 se află pe cromozomul X și determină tipul 1. Bărbații au un singur cromozom X. Astfel, dacă cromozomul X prezintă mutația genei KAL1, bărbații vor avea boala. Femeile prezintă 2 cromozomi X, iar pentru a avea boala este necesară mutația genei KAL1 în ambii cromozomi. O caracteristică a bolilor cu transmitere legată de cromozomul X este că bărbații cu sindrom Kallmann nu pot transmite boala fiilor lor. [1]
În general, persoanele moștenesc mutația de la mamă, care are o singură mutație. Există și posibilitatea unei mutații noi a genei. [1]
Alte forme de sindrom Kallmann pot fi moștenite autozomal dominant, ceea ce înseamnă că o singură mutație este suficientă pentru a cauza boala. Transmisia poate fi de la un părinte bolnav. Uneori, există o mutație nouă a genei, fără istoric de sindrom Kallmann în familie. [1]
O altă modalitate de transmisie a sindromului este autozomal recesiv. Transmiterea autozomal recesivă necesită două mutații (în ambii cromozomi). În acest caz, ambii părinți au câte o mutație, fără a avea semne clinice de boală. [1]
Diagnostic
La copii, este normal ca nivelul hormonilor sexuali să fie scăzut. Pentru diagnosticul sindromului Kallmann, se realizează un test de stimulare. Testul constă în administrarea de GnRH injectabil, fiind urmată de măsurarea nivelului hormonilor sexuali. În sindromul Kallmann hormonii sexuali rămân scăzuți după stimulare. [3]
Diagnosticul sindromului Kallmann necesită teste genetice, pentru a identifica mutația genei responsabile. Această testare se face prin recoltarea a 5-10 ml de sânge periferic. Rezultatul testării este disponibil, în general, după 6 săptămâni. [2]
În cazul în care există mutația în familie, poate fi indicat diagnosticul antenatal. [2]
Tratament
Dacă persoanele cu sindrom Kallmann nu beneficiază de tratament, acestea rămân infertile și nu pot avea copii. [3]
Pentru inducerea și menținerea caracterelor sexuale secundare, este recomandată administrarea de steroizi gonadali. În cazul bărbaților se vor administra testosteron sau hCG (gonadotropina corionică umană) injectabil. Femeile beneficiază de tratament cu estrogen. Dozele vor fi crescute progresiv. [2]
Testosteronul contribuie la creșterea și dezvoltarea în timpul pubertății. Un alt scop al terapiei hormonale este asigurarea mineralizării osoase. Astfel, riscul de osteoporoză este scăzut. [3]
Pentru stimularea spermatogenezei și a foliculogenezei (inducerea ovulației) se poate combina hCG cu hMG (gonadotropina menopauzală umană) sau terapie cu GnRH pulsatil. În caz de eșec al terapiei, există posibilitatea fertilizării in vitro. [2]
Terapia hormonală este de lungă durată. [3]
Nu există terapie pentru a corecta scăderea simțului olfactiv. [3]
De reținut
- Sindromul Kallmann este caracterizat prin lipsa simțului mirosului și prin tulburări de maturizare sexuală.
- Sindromul Kallmann este o afecțiune genetică, mai frecvent întâlnit la sexul masculin. În lipsa tratamentului, copilul cu sindrom Kallmann nu va trece prin pubertate și nu va putea face copii. Terapia hormonală de lungă durată este necesară pentru o creștere și dezvoltare normală.
- De asemenea, terapia este necesară pentru mineralizarea osoasă și prevenția osteoporozei.
- Nu există tratament pentru deficitul simțului olfactiv.
- Helicobacter pylori
- Colagenoza nespecifica. Polimiozita
- Ganglioni axilari inflamati
- Urticarie colinergica
- Boala Behcet
- Hiv sau paranoia?
- Alergie la toti alergenii posibili
- Ce vaccin sau tratament pot face sa-mi intaresc imunitatea
- Exista vreo sansa sa NU am lupus...?
- Ganglioni limfatici inflamati de doi ani de zile