Sindromul Fanconi
©
Autor: Istratie Elena-Bianca
Fiziopatologie
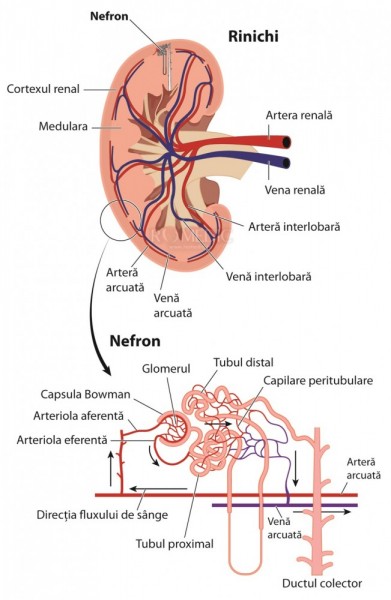
La nivelul tubului contort proximal se reabsorb cele mai multe substante filtrate fiziologic : albumina, aminoacizii, glucoza, bicarbonatul, sodiul, cloru, fosfatul, acidul uric. Aceste procese de reabsortie se realizeaza prin endocitoza mediata de proteinele megalina/cubilina si transportori sodiu dependenti. Procesul de endocitoza este foarte important pentru reabsorbtie si pentru degradarea proteinelor care traverseaza bariera de filtrare glomerulara. Proteinele care trec filtrul glomerular (albumine, proteine cu greutate moleculara mica, hormoni, proteine care leaga vitaminele) sunt reabsorbite prin procese de endocitoza mediate de megalina si cubilina (receptori implicati in procesul de endocitoza) de la nivelul membranei luminale. Apoi complexul proteina-receptor este incorporat in endozom. In endozom se produce disocierea receptorului de proteina. Receptorul este reciclat in membrana luminala iar produsii de reabsorbite sunt procesati in lizozomi. Aceasta disociere a receptorului de produsul de reabsortie are loc intr-un mediu acid dependent de concentratia de H+ si Cl- din endozom, concentratie mentinuta de ATP-aza de H+ (de protoni) si de canalul de clor ClC-5.Alterarea procesului de endocitoza afecteaza reciclarea proteinelor transportoare (megalina si cubilina) catre membrana luminala determinand diminuarea reabsortiei. Astfel se vor pierde in urina cantitati insemnate de albumina, proteine cu greutate moleculara mica, electroliti, . Cadmiul inhiba ATP-aza de protoni si mitocondria, producand un sindrom asemanator sindromului Fanconi (Fanconi-like syndome). Folimicinul, un alt inhibitor al ATP-azei de protoni, impiedica reabsortia albuminei din tubul contort proximal. In boala Dent, afectiune caracterizata printr-un defect al canalului de clor ClC-5, sunt prezente manifestari similare celor din sindromul Fanconi, secundare alterarii procesului de reciclare a megalinei la nivelul membranei luminale. In analiza urinei normale se gaseste megalina. La pacientii cu boala Dent sau la cei cu sindrom Lowe, megalina este absenta in urina.
Reabsortia solventilor (glucoza, fosfati, aminociazi, bicarbonat) din tubul contort proximal se realizeaza printr-un sistem transportor de la nivelul membranei care este dependent de Na+, de energia mitocondriala si de ATP-aza Na+ si K+ din membrana bazo-laterala. In cazul in care este alterata productia de energie necesara functionarii acestor pompe, este afectat transportul la nivelul tubului contort proximal.
Desi cei doi rinichi cantaresc mai putin de 1% din greutatea corpului, consuma 10% din energia totala in repaus. Cea mai mare parte a energiei este folosita la nivelul tubului contort proximal in procesele de transport. Cand se produc defecte de generare a energiei, in tubul contort proximal este alterat transportul produsilor de reabsortie, fenomen care determina sindromul Fanconi caracterizat prin pierderi urinare de aminoacizi, glucoza, fosfat, bicarbonat, acid uric.
Peste 13000 de cabinete medicale își prezintă serviciile pe ROmedic.
Etiologie
Sindromul Fanconi este trei feluri: ereditar, dobandit si produs de substante exogene.Tipul ereditar este consecinta unor anomalii metabolice congenitale. Forma dobandita este determinata de reactii imune, de sindromul nefrotic sau in urma acumularii de proteine anormale. Substantele exogene raspunzatoare de sindromul Fanconi sunt: medicamente, substante chimice, metale grele.
Sindromul Fanconi de natura ereditara
Boala Dent
Este o tubulopatie proximala X-linkata, caracterizata prin albuminurie, hipercalciurie, nefrocalcinoza, nefrolitiaza, pana la insuficienta renala. Unii pacienti pot prezenta rahitism si acidoza metabolica. Cei multi pacienti sunt barbati. Adultii cu boala Dent prezinta sindromul Fanconi, iar copiii prezinta microalbuminurie si cateva din manifestarile tubulopatiei proximale care realizeaza un sindrom Fanconi partial. Acestia pot ajunge la insuficienta renala la varsta de 40 de ani. Totusi o treime din pacientii cu boala Dent nu dezvolta insuficienta renala. Singura manifestare extrarenala la acesti pacienti este rahitismul secundar fosfaturiei.Boala Dent este determinata de mutatia genei CLCN5 care codifica informatia pe baza careia se codifica aminoacizii canalului 5 de clor (ClC-5) . Acest canal de clor este asociat cu ATP-aza H+ avand rol in acidifierea endozomului in scopul transportului proteinelor. In aceasta afectiune sunt descrise peste 80 de mutatii ale CLCN5. Disfunctia canalului de clor Cl-C5 determina perturbarea procesului de endocitoza prin impiedicarea disocierii magalinei si cubilinei, scazand expresia acestor receptori in membrana luminala ceea ce are drept consecinta diminuarea reabsortiei cu albuminurie, hipercalciurie, hiperfosfaturie si nefrolitiaza. Mediul acid endozomal este important si pentru degradarea proteinelor.
Nivelul urinar al proteinelor este de 0,5-2,5 g/zi ajungand pana la 4 g/ zi. Mai mult de 60% din proteinele filtrate sunt proteine cu greutate moleculara mica (albumina, β2-microglobulina). Nu se instaleaza sindromul nefrotic. Acesti pacienti prezinta nefrocalcinoza si calciurie care in timp duc la scaderea ratei filtratului glomelular si insuficienta renala in jurul varstei de 40 de ani. Biopsia renala evidentiaza glomeruloscleroza focala, atrofie tubulara, dilatare tubulara, infiltrat interstitial cu monocite. Nefrocalcinoza medulara este caracteristica pentru aceasta afectiune.
Tratamentul cu inhibitori ai enzimei de conversie ai angiotensinei (IECA) sau cu blocanti ai receptorului pntru angiotensina (ARB) intarzie progresia catre insuficienta renala.
Sindromul Lowe (sindromul oculo-cerebro-renal OCRL)
Este o afectiune X-linkata in care se produc anomalii ale ochilor (cataracta congenitala), ale sistemului nervos central si apare sindromul Fanconi. Au fost descrise mai mult de 70 de mutatii ale genei OCRL1 la pacientii cu sindrom Lowe. Toti pacientii de sex masculin prezinta cataracta. Alte afectiuni oculare sunt: glaucomul, microftalmos, formarea de cheloid la nivelul corneei. Acuitatea vizuala este alterata. Afectarea sistemului nevos central determina hipotonie infantila, abolirea reflexelor, retard mental. Pacientul prezinta ventriculomegalie si chiste periventriculare. Uneori se instaleaza status epilepticus (criza epileptica cu durata de peste 30 de minute sau mai multe crize fara recapatarea completa a constientei intre ele durand peste 30 de minute).Sindromul Fanconi apare inca din primul an de viata. Pacientul prezinta albuminurie, glucozurie, aminoacidurie, hiperfosfaturie, hipercalciurie, rahitism hipofosfatemic, acidoza metabolica hipercloremica, insuficienta renala progresiva.
In sindromul Lowe precum in sindromul Dent, megalina este aproape absenta din urina, sugerand expresia redusa a megalinei la nivelul membranei luminale a tubului contort proximal.
Tratamentul este suportiv vizand ameliorarea manifestarilor oculare, anticonvulsivante, logopedie, complicatiile dentare. Pentru corectarea acidozei se administreaza bicarbonat. De asemenea pacientul primeste sodiu, potasiu, fosfat, vitamina D.
Mitocondriopatii
Mitocondria este un organit celular care indeplineste multiple sarcini: oxidarea acizilor grasi, rol in ciclul acizilor tricarboxilici, ciclul ureei, producerea de ATP prin procesul de fosforilare oxidativa.Fosforilarea oxidativa are loc in cadrul lantului respirator de la nivelul membranei interne mitocondriale. Lantul respirator are cinci componente: complexul I (NADH-coenzima Q reductaza), complexul II (succinat-coenzima Q reductaza), complexul III (coenzima Q-citocrom c reductaza), complexul IV (citocrom c oxidaza) si complexul V (ATP sintetaza). Fosfoilarea oxidativa cuprinde reactii de oxidare care consuma oxigen si prin care ADP in urma fosforilarii devine ATP. ADN-ul fiecarei mitocondrii prezinta 37 de gene: 13 codifica polipeptidele lantului respirator, 2 codifica ARN-ul ribozomal, 22 pntru ARN-ul de transfer. Lezarea mitocondriei poate fi congenitala sau secundara. Afectarea genetica a ADN-ului mitocondrial determina mitocondriopatii.
Mitocondriopatiile sunt afectiuni multisistemice care se pot manifesta la orice varsta. Sunt afectate multiple organe: sistemul nervos central, muschiul, ficatul, inima, rinichii, intestinele, sistemul endocrin, maduva osoasa, urechea, ochiul, pielea.
Investigatiile specifice acestei afectiuni includ: lactatul plasmatic, piruvatul, corpii cetonici, studii spectrometrice pentru evaluarea complexelor enzimatice al lantului respirator, biopsie musculara si analiza genetica.
Afectarea renala poate fi prima manifestare a mitocondriopatiei sau se poate manifesta concomitent cu afectarea neurologica si neuro-musculara. Sindromul Fanconi este mult mai frecvent in acest caz la nou-nascuti si la copiii mici, la care nefropatia tubulo-interstitiala este asociata cu glomeruloscleroza facala segmentara ereditara.
Multi dintre pacientii cu sindrom Fanconi in contextul mitocondriopatiei prezinta deficit de crestere, aminoacidurie, glucozurie, proteinurie, uricosuria, hipercalciurie si bicarbonaturie. Acesti pacienti dezvolta sindromul Fanconi de la varsta de doi ani.
Manifestarile extrarenale includ simptome neurolgice, miopatie, afectare hepatica, trasaturi clinice ale sindromului Pearson, insuficienta suprarenala partiala, afectare cardiaca, diabet zaharat, surzenie, oftalmoplegie. Unii dintre pacienti evolueaza catre insuficienta renala. La examenul histopatologic se evidentiaza dilatatii tubulare, atrofie tubulara, mitocondrii gigante.
Nu exista tratament curativ pentru mitocondropatii. Se administreaza tratament simptomatic: suplimente de bicarbonat de sodiu, potasiu, vitamina D3, fosfat, apa. In cazul deficitului de carnitina se administreaza carnitina. Este recomandata evitarea medicamentelor care interfera cu lantul respirator precum: valproatul, barbituricele sau a celor care inhiba sinteza proteinelor mitocondriale: tetraciclina, cloramfenicolul. Pentru pacientii cu defect al complexului I este recomandata o dieta care include grasimi in cantitate mai mare si putini carbohidrati.
Cistinoza
Este a afectiune autozomal recesiva in care stocarea in lizozom este alterata cu acumularea de cistina in organe ca rinichiul, cornee (fotofobie), maduva osoasa, tiroida (hipotiroidism), noduli limfatici, ficat si splina. In taril vestice cistinoza este cea mai frecventa afectiune responsabila de sindromul Fanconi.Cistinoza infantila este cea mai frecventa forma si se manifesta prin deficit de crestere, poliurie, polidipsie, deshidratare, pierderi hidro-electrolitice, aminoacidurie, glucozurie, fosfaturie, acidoza renala tubulara in jurul varstei de 6-12 luni. Unii dintre pacienti pot dezvolta rahitism rezistent la vitamina D secundar fosfaturiei, cu deficit de crestere insemnat. In absenta tratamentului acesti copii pot ajunge la insuficienta renala in jurul varstei de 10 ani.
La examenul hitopatologic se evidentiaza leziuni ale tubilor contorti proximali, leziuni ale podocitelor (devin celule gigante multinucleate) si prezenta cristalelor de cistina in celulele interstitiale si in cele podocitare. Depunerea cristalelor de cistina in cornee determina fotofobie.
Alte doua forme mai putin severe in cistinoza sunt cistinoza juvenila si oculara. Pacientii cu cistinoza juvenila prezinta leziuni glomerulare in jurul varstei de 12-15 ani; acestia au o crestere normala. Cei cu cistinoza oculara nu au afectare renala.
Tratamentul cistinozei infantile include refacerea balantei hidro-electrolitice. Se administraeaza cisteamina (chelator de cistina) per os, medicament care incetineste progresia spre insuficienta renala, amelioreaza semnificativ functia tiroidiana, indeparteaza cistina din muschi. Cisteamina se administreaza in continuare dupa transplantul renal pentru imbunatatirea functiei celorlalte organe afectate. Administrarea de cisteamina sub forma de picaturi pentru ochi determina dizolvarea cristaleler de cistina de la nivelul corneei si amelioreaza functia vizuala. Totodata, terapia cistinozei include si potasiu, agenti alcalinizanti (citrat sau bicarbonat), fosfat si vitamina D3. In cazul in care pacientul prezinta deficit de crestere dupa un an de terapie, se face terapie cu hormon de crestere.
Galactozemia
Este o afectiune autozomal recesiva in care este alterat metabolismul galactozei. Galactoza este sursa de carbohidrati preferata de nou-nascuti. Galactozemia este rezultatul activitatii deficitare a fosfatazei-uridil-transferazei galactozei 1, enzima care catalizeaza reactia de fosforilare a galactozei 1 (gal-1-p) . Laptele este principala sursa de galactoza. In galactozmie se acumuleaza gal-1-p din cauza activitatii deficitare a GALT, iar expunerea la galactoza determina afectarea acuta a numeroase organe: ficat, rinichi, ovare, creier, ochi.Copiii se prezinta pentru voma, diaree, stagnare ponderala, disfunctie hepatica, coagulopatie, disfunctie tubulara renala, edem cerebral, sepsis cu Echerichia Coli. Uneori au icter cu hiperbilirubinemie neconjugata si hemoliza sevara. Afectarea hepatica se poate complica cu hepatomegalie si ciroza care poate determina moartea. Este important diagnosticul precoce si instalarea dietei de restrictie a galactozei in scopul prevenirii complicatiilor pe termen lung precum retardul mental, insuficienta ovariana, tulburarile de vorbire.
Rinichii sunt si ei afectati, principala manifestare fiind sindromul Fanconi in care apar: hiperaminoacidurie, albuminurie, hiperfosfaturie, bicarbonaturie.
Pacientii care tin dieta de restrictie a galactozei, nu sunt complet privati de efectele galactozei, intrucat aceasta se gaseste in vegetale si fructe. Pe de alta parte galactoza este sintetizata si pe cale endogena. Insa acesti pacienti nu manifesta complicatiile cronice intalnite la pacientii cu galactozemie clasica. Ei prezinta cataracta.
Tratamentul vizeaza eliminarea galactozei din dieta. Simptomele se remit in cateva zile.
Intoleranta ereditara la fructoza
Este o afectiune autozomal recesiva determinata de deficitul de aldolaza B, enzima care catalizeaza metabolizarea fructozei exogene la nivelul ficatului, intestinului si corticalei renale. Indivizii afectati prezinta hipoglicemie, varsaturi, le poate fi amenintata viata dupa ingestia de fructoza, sucroza sau sorbitol. Ingestia prelungita va duce la deficit de crestere, hepatomegalie, icter, ciroza hepatica si nefrocalcinoza, convulsii, coma, chiar moartea secundara insuficientei hepatice si renale.Simptomele apar in copilarie. Copiii manifesta aversiune fata de produsele care contin fructoza: dulciuri, fructe.
Intoleranta ereditara la fructoza se asociaza cu disfunctie de tub contort proximal care conduce la aminoacidurie, bicarbonaturie, fosfaturie, acidoza lactica. Ingestia cronica de fructoza determina nefrocalcinoza.
Tratamentul consta in evitarea alimentelor sau medicamentelor care contin fructoza, sucroza sau sorbitol.
Boala de stocare a glicogenului de tip I (von Gierke)
Este o afectiune autozomal recesiva care are o incidenta de de 1 la 100 000 de locuitori. Are doua subtipuri: Ia este cauzata de deficitul de glucozo-6-α-fosfataza (catalizeaza hidroliza glucozo-6-fosfatazei la glucoza) si Ib cauzata de deficitul de transportor al glucozo-6-fosfataza.Pacientii cu GSD-Ia prezinta dezechilibre ale metabolismului glucidic care se manifesta prin hipoglicemie, hepatomegalie, nefromegalie, hipercolesterolemie, hipertrigliceridemie, hiperuricemie, acidoza lactica, neutrofilie, deficit de crestere. Aproximativ 75% din adolescentii si adultii cu aceasta afectiune dezvolta adenom hepatocelular. Complicatiile renale includ hipertrofia renala, calculi renali, guta secundara nefropatiei, nafrocalcinoza, sindromul Fanconi-like, insuficienta renala cronica.
Tratamentul are ca obiective principale normalizarea glicemiei si tratamentul acidozei lactice. Normoglicemia este atinsa prin administrarea glucozei pe sonda nazo-gastrica. Transplantul hepatic este indicat cand nu se mai realizeaza controlul metabolic prin tratamentul medicamentos sau cand se dezvolta adenom hepatocelular sau carcinom hepatocelular.
Sindromul Fanconi-Bickel
Este o afectiune autozomal recesiva caracterizata prin deficit de crestere, hepatomegalie, nefromegalie, fata „de papusa”, rahitism. Acesti pacienti prezinta acumulare de glicogen la nivelul hepatocitelor si celulelor tubului contort proximal, hipoglicemie, intoleranta la galactoza si manifestarile sindromului Fanconi. Unii dintre pacienti prezinta cataracta neonatala, altii au diabet zaharat neonatal si galactozemie.Acest sindrom se produce in urma unor mutatii la nivelul genei transportorului de glucoza care se gaseste in ficat, rinichi, intestin si in insula pancreatica.
Tratamentul include bicarbonat de sodiu, fosfat de potasiu, tratamentul rahitismului cu vitamina D3, alimentare perioada noptii pentru a preveni cetoacidoza. Este preferat laptele fara galactoza.
Tirozinemia I
Este o afectiune autozomal recesiva in care este elterat metabolismul aminoacidului tirozina. Este consecinta unui defect in gena hidrolazei fumarilacetoacetatului (FAH) . FAH este ultima enzima din catabolismul tirozinei.Acesti pacienti prezinta afectare hepatica cu debut in copilarie care evolueaza spre ciroza, deficit al factorilor de coagulare, hipoglicemie, concentratii plasmatice crescute de metionina, fenilalanina si acid aminolevulinic, risc crescut de carcinom hepatocelular si afectare renala tubulara si glomerulara.
Boala Wilson
Este o afectiune autozomal recesiva in care este alterat metabolismul cuprului Cu. Sunt afectate numeroase organe. Este afectata excretia biliara a Cu si legarea de ceruloplasmina ceea ce determina afectare hepatica, degenerare neuronala.Aproximativ 40% din pacienti se prezinta cu afectare hepatica, 40% cu manifestari extrapiramidale, 20% cu tulburari de comportament si psihice.
Acumularea in exces a Cu la nivelul rinichilor conduce la disfunctie tubulara renala. Manifestarile sindromului Fanconi la acesti pacienti se instaleaza inaintea simptomelor hepatice. Ei prezinta glucozurie, aminoacidurie, hipefosfaturie, hiperuricozurie, proteinurie. De asemenea pot avea rahitism, osteomalacie, hipercalciurie, litiaza renala, nefrocalcinoza.
Terapia are scopul indepartarii Cu din organism, folosind chelatori de Cu (D-penicilamina) si evitarea alimentelor ce contin Cu: ciocolata, nuci, ciuperci, ficat.
Sindromul Fanconi dobandit
Sindromul nefrotic este asociat cu sindromul Fanconi. Glomeruloscleroza segmentara poate determina sindrom nefrotic.Afectiuni imune sau hematologice sunt asociate cu proteinurie: mielomul multiplu, sindromul Sjӧgren, amiloidoza. In faza precoce a mielomului multiplu pacientii prezinta afectarea tubilor renali proximali care determina sindrom Fanconi. La pecientii cu sindrom Sjӧgren (afectiune a tesutului conjunctiv) se deceleaza proteinurie, iar 4% dintre ei manifesta sindrom Fanconi.
Nefrita tubulo-interstitiala acuta cu uveita este o afectiune imuna in care pacientii manifesta astenie, maleza (stare de rau), scadere ponderala, sete, sindrom Fanconi. Corticoterapia amelioreaza simptomatologia renala si oculara.
In nefrita autoimuna interstitiala si in nefropatia membranoasa pacientii manifesta deficit de crestere, afectare renala tubulara care conduce la proteinurie si sindrom Fanconi.
Sindromul fanconi produs de substante exogene
Numeroase medicamente si plante eliminate pe cale renala sunt responsabile de sindromul Fanconi. Astfel tetraciclina expirata, aminoglicozidele, salicilatii, acidul valproic si ierburile chinezesti pot altera filtrul renal. Aminoglicozidele (antibiotice) scad reabsortia glucozei. Salicilatii altereaza functia mitocondriei celulelor tubilor proximali. Acidul valproic produce anomalii de transport la nivelul celulelor tubulare proximale in urma afectarii lantului respirator mitocondrial. Ierburile chinezesti contin acid aristolohic care produce leziuni tubulare proximale.Chimioterapicele produc disfunctie renala tubulara si glomerulara, inclusiv sindrom Fanconi. Nefrotoxicitatea acestor medicamente este dependenta de doza si uneori ireversibila. Chloroacetaldehida (metabolit al Ifosfamidei) inhiba endocitoza in celulele tubulare proximale. Cisplatin reduce reabsortia glucozei si a numerosi aminoacizi. Imanitib mesilat folosit in tratamentul leucemiei cronice mieloide induce sindrom Fanconi.
Inhibitorii nucleotidici de revers transcriptaza folositi in tratamentul HIV (adefovir, cidofovir, tenofovir) determina sindrom Fanconi, diabet insipid, insuficienta renala acuta.
Metalele grele: plumb, cadmiu, mercur, cromiu, platina sunt foarte toxice de la doze mici. Rinichiul este principalul organ afectat. Dupa intoxicatia cu plumb, aninoaciduria si glicozuria persista aproape 13 ani.
Examenul clinic
Deficitul de crestere este trasatura comuna a copiilor cu sindrom Fanconi. Apare in urma malnutritiei, hipopotasemiei, hipofosfatemiei si acidozei metabolice. Deficitul de potasiu isi lasa amprenta asupra procesului de crestere fiind in relatie directa cu hormonul de crestere si hormonul (GH) de crestere insulin-like (IGF-1) ale caror concentratii scad. Hipopotasemia altereaza apetitul determinand malnutririe. Acidoza metabolica inhiba expresia receptorilor pentru GH si IGF-1. Hipofosfatemia afecteaza oasele determinand rahitism si deficit de crestere. Pacientii se prezinta cu dureri osoase si articulare.La pacientii cu sindrom Fanconi sunt intalnite poliuria, polidipsia si deshidratarea. Poliuria este generata de diureza osmotica. La copiii de 6-12 luni cu sindrom Fanconi apare febra acuta recurenta care produce deshidratare.
Investigatii de laborator
Aminoaciduria este prezenta la pacientii cu sindrom Fanconi. Aproximativ 95-99% din aminoacizi se reabsorb la nivelul tubului contort proximal.Glucozuria apare in sindromul Fanconi in urma afectarii reabsortiei glucozei. Zilnic se pierd in urina 0,5-20g de glucoza in urina.
Hipofosfatemia duce la rahitism si osteomalacie prin afectarea procesului de hidroxilare a vitaminei D3 in celulele tubulare proximale.
Acidoza metabolica este secundara alterarii reabsortiei bicarbonatului care se reabsoarbe in conditii fiziologice in proportie de 85% la acest nivel.
Sodiul si potasiul se pierd in urina. Hiponatremie induce hipotensiune si deshidratare. Hipopotasemia severa poate determina moartea.
Hipercalciuria apare in urma endocitozei defectuoase a parathormonului la pacientii cu boala Dent.
Hiperuricozuria este prezenta. 90-95% din acidul uric este reabsorbit la nivel proximal.
Proteinuria se refera la pierderi pe cale urinara de albumina si gamaglobuline.
Tratament
Primul pas in initierea tratamentului il constituie determinarea agentului declansator al sindromului Fanconi.In galactozemie, intoleranta ereditara la fructoza si tirozinemie se inlatura alimentele care contin aceste elemente. In boala Wilson se evita alimentele bogate in cupru si de administreaza chelatori de cupru (D-penicilamina, trientina, amoniu tetratiomolibdat) si zinc.
Medicamentele imunosupresoare se folosesc pentru afectiunille de natura imuna (sindromul sjӧgren, sindromul TINU, nefrita interstitiala autoimuna, nefropatia membranoasa).
Pentru acidoza tubulara proximala se administreaza substante alcalinizante (saruri de potasiu, citrat, bicarbonat) . Pentru deficitul de sodiu si deshidratare sunt indicate bicarbonatul de sodiu, citrat, clorura, in functie de gradul de acidoza. Rehidratarea este esentiala.
Profilaxia rahitismului si osteomalaciei se face cu 1,25-dihidroxi-vitamina D3. Totodata se administreaza fosfat. Preventia deformarilor osoase si a fracturilor se face cu calciu, fosfat si vitamina D3. Pacientilor cu deficit de crestere important li se administreaza hormon de crestere.
Din Biblioteca medicală vă mai recomandăm:
- Abcesul perinefretic si renal
- Diabet insipid
- Sindromul Alport - Nefrita ereditara
- Abcesul renal
- Glomerulonefrita
- Insuficienta renala acuta - IRA
- Insuficiența renală cronică - IRC
- Litiaza renala
- Rinichi polichistic
- Sindromul nefrotic
- Miros anormal al urinei
- Mancarea fast food si disfunctiile renale - exista o legatura?
- Rinichii
Din Ghidul de sănătate v-ar putea interesa și:
Forumul ROmedic - întrebări și răspunsuri medicale:
Pe forum găsiți peste 500.000 de întrebări și răspunsuri despre boli sau alte subiecte medicale. Aveți o întrebare? Primiți răspunsuri gratuite de la medici.- Ajutor: cancer renal metastaze la ficat
- Dureri puternice in dreapta buricului
- Un singur rinichi si imporsibilitate de a urina
- Durere rinichi
- Ecografie abdominala nodul si chist rinichi
- Transplant de rinichii
- Rinichi bolnav, senzatia de intoxificare, stare de somnolenta
- Rinichi drept dilatat
- Pot sa raman insarcinata cu un singur rinichi?
- Lovitura rinichi